Siguiente: Acoplamiento mecánico con el Subir: Ensamble canónico Anterior: Modelo de Debye
El análisis de las moléculas diatómicas resulta de especial interés porque permite avanzar en la comprensión de diferentes situaciones. Para ello, consideramos las moléculas de un gas diatómico como rotadores rígidos, conformados por dos átomos puntuales mantenidos a distancia fija. A temperatura ambiente, los grados de libertad electrónicos y de vibración no están excitados, de manera que esta aproximación resulta muy adecuada.
Los grados de libertad traslacionales se corresponden con los de una
partícula cuántica en una caja (tridimensional) de lado ![]() , de modo que los
niveles de energía estarán dados por
, de modo que los
niveles de energía estarán dados por
![]() (
(
![]() ). Para una masa atómica del
orden de 10
). Para una masa atómica del
orden de 10![]() g y una caja de lado
g y una caja de lado ![]() cm, la energía térmica
relacionada con el salto entre dos niveles consecutivos de energía
corresponde a una temperatura de 10
cm, la energía térmica
relacionada con el salto entre dos niveles consecutivos de energía
corresponde a una temperatura de 10![]() K, de modo que a temperatura
ambiente, la contribución de los modos traslacionales a la energía interna
puede calcularse mediante un enfoque clásico.
K, de modo que a temperatura
ambiente, la contribución de los modos traslacionales a la energía interna
puede calcularse mediante un enfoque clásico.
Nos concentramos entonces en la energía cinética de rotación, cuya expresión
clásica, en términos del momento de inercia ![]() es
es
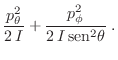

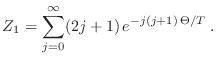
Para el caso
![]() , podemos hacer la sustitución
, podemos hacer la sustitución
![]() ,
resultando una variable continua en este rango de temperaturas. De este modo, obtenemos
,
resultando una variable continua en este rango de temperaturas. De este modo, obtenemos

Así, para la energía media por molécula
![]() encontramos:
encontramos:
![$\displaystyle c_v =
k \left(\displaystyle\frac{\Theta}T\right)^2
\left[ 12 e^{-2\Theta/T} - 72 e^{-4\Theta/T} + \dots \right]$](img689.png) |
|
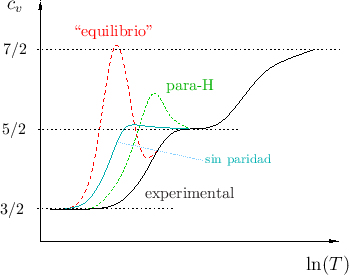
Para el caso de la molécula de hidrógeno, los valores predichos para
En cambio, lo que no se ha tenido en cuenta es el espín 1/2 de los núcleos
de hidrógeno, ni tampoco el hecho de que el espín total
|
|
Cuando los átomos son distintos, en muy buena aproximación, la degeneración de
cada estado no influye en el valor de la energía, porque esta será casi
independiente de ![]() . En ese caso, la función partición debe
multiplicarse por un factor constante
. En ese caso, la función partición debe
multiplicarse por un factor constante
![]() (
( ![]() en nuestro
caso).
en nuestro
caso).
Sin embargo, cuando los átomos son idénticos, debe tenerse en cuenta la indistinguibilidad de las partículas. El postulado de simetrización
exige que la función de onda conjunta de la molécula sea antisimétrica
al intercambiar los átomos. Las partes correspondientes a vibración y a
estados electrónicos son simétricas en el estado base (no están excitadas).
En el para-estado, la función de onda de espín es antisimétrica, mientras
que en el orto-estado es simétrica. La función de onda rotacional es
simétrica o antisimétrica según el número cuántico ![]() sea par o impar
11. O sea que tendremos:
sea par o impar
11. O sea que tendremos:
| para-hidrógeno (
|
|
degeneración 1; |
| orto-hidrógeno (
|
|
degeneración 3. |
Así, la función partición debe calcularse teniendo en cuenta que debe sumarse sobre todos los estados posibles:
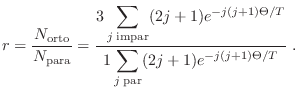
Hay otras alternativas para verificar que el cálculo mediante este método es
correcto. Una de ellas es medir el calor específico de mezclas con
diferentes valores para ![]() . Para hacerlo, el gas se prepara pasándolo por
un catalizador (carbono activado) a diferentes temperaturas, con el objeto de
acelerar la transición orto-para: en esos experimentos las predicciones concuerdan con los datos medidos.
. Para hacerlo, el gas se prepara pasándolo por
un catalizador (carbono activado) a diferentes temperaturas, con el objeto de
acelerar la transición orto-para: en esos experimentos las predicciones concuerdan con los datos medidos.
Las mismas consideraciones que hemos hecho valen para el deuterio diatómico
D![]() . En el caso del HD, por supuesto, no surgen inconvenientes, ya que los
dos átomos son diferentes y las cuentas con que comenzamos eran válidas.
. En el caso del HD, por supuesto, no surgen inconvenientes, ya que los
dos átomos son diferentes y las cuentas con que comenzamos eran válidas.
Gustavo Castellano 19/11/2021