Siguiente: Discontinuidad en la entropía Subir: clases-t1 Anterior: Principio de Le Châtelier-Braun
Así como el agua líquida a presión atmosférica se solidifica al descender su temperatura por debajo de 273,15 K y se evapora al calentarse por encima de 373,15 K, los sistemas termodinámicos en general pueden existir en diferentes fases: en las temperaturas de transición ocurren cambios abruptos en algunas propiedades macroscópicas.
En general, los sistemas se vuelven menos desordenados a medida que desciende la temperatura: en el caso de un fluido, las fuerzas de cohesión comienzan a ganarle al movimiento térmico, de modo que los átomos se acomodan en estados regidos por cierta regularidad. En el contexto de la termodinámica, sólo nos concentramos en las variables macroscópicas; tal como anticipamos en secciones anteriores, cada transición de fase está asociada con una región lineal en la relación fundamental, que corrige alguna falla en el criterio de estabilidad termodinámica.
Si bien en la coexistencia cada fase tiene sus propiedades termodinámicas
bien definidas (por ejemplo, ![]() y
y ![]() constantes), los subsistemas
(fases) pueden intercambiar materia al estar en equilibrio, por lo que
sabemos que debe cumplirse que los potenciales químicos de cada fase deben
ser iguales. Esto significa que tanto
constantes), los subsistemas
(fases) pueden intercambiar materia al estar en equilibrio, por lo que
sabemos que debe cumplirse que los potenciales químicos de cada fase deben
ser iguales. Esto significa que tanto ![]() como
como ![]() deben cambiar continuamente al ocurrir una transición de fase. Las transiciones se
clasifican según la continuidad de las derivadas del potencial de Gibbs:
cuando hay cambios de estado discontinuos (primeras derivadas de
deben cambiar continuamente al ocurrir una transición de fase. Las transiciones se
clasifican según la continuidad de las derivadas del potencial de Gibbs:
cuando hay cambios de estado discontinuos (primeras derivadas de ![]() discontinuas), tenemos una transición de fase de primer orden y los
estados correspondientes a cada fase se hallan en regiones separadas del
espacio de configuraciones termodinámicas; cuando los cambios de estado son
continuos (derivadas superiores discontinuas), nos encontramos con transiciones de fase continuas o de orden superior, también
denominados fenómenos críticos.
discontinuas), tenemos una transición de fase de primer orden y los
estados correspondientes a cada fase se hallan en regiones separadas del
espacio de configuraciones termodinámicas; cuando los cambios de estado son
continuos (derivadas superiores discontinuas), nos encontramos con transiciones de fase continuas o de orden superior, también
denominados fenómenos críticos.
En la discusión que prosigue, a menudo invocaremos el caso de los fluidos clásicos, pues conforman uno de los ejemplos más familiares de transiciones de fase de primer orden.
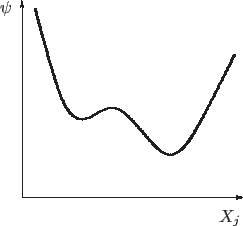
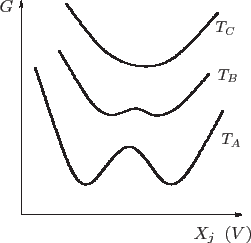
Para comprender mejor la dinámica de una transición fijamos la atención en
la energía libre de Gibbs, que para cierta temperatura presenta dos mínimos,
uno local y el otro global. En la gráfica que se muestra a continuación, las
variables naturales de ![]() se encuentran fijas y estudiamos las
variaciones a causa de algún otro parámetro
se encuentran fijas y estudiamos las
variaciones a causa de algún otro parámetro ![]() , relacionado con la
cantidad de sistema en cada fase. Por ejemplo, en el caso de un fluido
clásico, mantenemos
, relacionado con la
cantidad de sistema en cada fase. Por ejemplo, en el caso de un fluido
clásico, mantenemos ![]() y
y ![]() constantes en un sistema cerrado, pero el
volumen total
constantes en un sistema cerrado, pero el
volumen total ![]() del sistema puede variar, ya que la densidad de cada
fase es diferente.
del sistema puede variar, ya que la densidad de cada
fase es diferente.
|
El sistema está en equilibrio para el valor de
|
|
Más adelante veremos que en el caso en que ![]() representa el momento
magnético, las transiciones involucran fases paramagnéticas y ferromagnéticas, cuyo estudio es motivo de numerosas investigaciones.
Cuando se analizan transiciones en las que se pasa de una estructura
cristalina a otra, el parámetro
representa el momento
magnético, las transiciones involucran fases paramagnéticas y ferromagnéticas, cuyo estudio es motivo de numerosas investigaciones.
Cuando se analizan transiciones en las que se pasa de una estructura
cristalina a otra, el parámetro ![]() relevante es una variable de
simetría, y resulta suficiente para caracterizar la transición.
relevante es una variable de
simetría, y resulta suficiente para caracterizar la transición.
|
Volviendo al gráfico anterior, si ahora reducimos la temperatura del
sistema, la forma de
Finalmente, a la temperatura
|
|
cionar que en este gráfico las ordenadas al origen correspondientes a cada temperatura no necesariamente coinciden.
En el ejemplo que habíamos sugerido con agua por encima de 373,15 K, si el vapor se enfría ``suavemente'' es posible seguir teniendo vapor aun por debajo de 373,15 K, es decir, puede pasarse de un estado estable a uno metaestable: en ese caso, cualquier fluctuación puede provocar que el sistema elija el mínimo global, que corresponde al estado líquido.
interpretar mejor las gráficas anteriores, conviene centrar la atención
en el caso particular del fluido clásico, en cuyo caso el parámetro ![]() es el volumen. Está claro que el sistema no pasa repentinamente de un volumen
correspondiente a la fase líquida a otro correspondiente a la gaseosa. El
hecho de que en un punto sean mínimos de
es el volumen. Está claro que el sistema no pasa repentinamente de un volumen
correspondiente a la fase líquida a otro correspondiente a la gaseosa. El
hecho de que en un punto sean mínimos de ![]() `equivalentes' significa que
cada elemento de masa del sistema puede elegir entre ambas fases, y eso es
lo que reconocemos como coexistencia. La combinación de diferentes
cantidades de sistema en cada fase debe respetar el volumen total
resultante, que debe coincidir con el de la vasija que lo contenga.
`equivalentes' significa que
cada elemento de masa del sistema puede elegir entre ambas fases, y eso es
lo que reconocemos como coexistencia. La combinación de diferentes
cantidades de sistema en cada fase debe respetar el volumen total
resultante, que debe coincidir con el de la vasija que lo contenga.
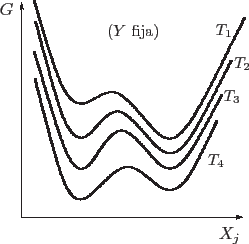
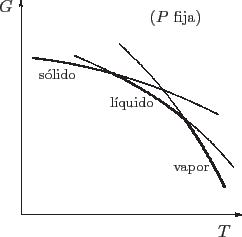
Una buena idea para avanzar con el análisis es graficar los mínimos que va
teniendo el potencial de Gibbs en cada fase como función de la temperatura,
siempre manteniendo fija la presión (o la variable ![]() para el caso
general). El equilibrio estable corresponde al mínimo valor posible para
para el caso
general). El equilibrio estable corresponde al mínimo valor posible para
![]() , de manera que la curva real es la envolvente inferior de esta
gráfica.
, de manera que la curva real es la envolvente inferior de esta
gráfica.
Vemos que aparecen discontinuidades en las derivadas de ![]() al cruzar las
temperaturas de transición. Recordando que
al cruzar las
temperaturas de transición. Recordando que
![]() , lo que estamos verificando en realidad es que la entropía es
discontinua. Esto es importante por dos razones: por un lado, esta
discontinuidad es la que caracteriza a la transición como de
, lo que estamos verificando en realidad es que la entropía es
discontinua. Esto es importante por dos razones: por un lado, esta
discontinuidad es la que caracteriza a la transición como de
|
primer orden; por otro lado, veremos en breve que este salto en la entropía se asocia con el calor latente de la transformación.
En estas representaciones faltaría una dimensión que corresponde a las
variaciones en la presión (
|
|
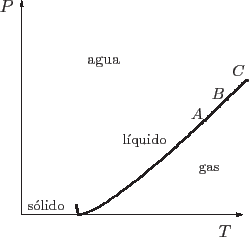
Cuando en una curva de coexistencia nos desplazamos hacia temperaturas mayores, podemos representar gráficos análogos a los anteriores teniendo en cuenta que estamos modificando ![]() y
y ![]() para seguir teniendo coexistencia de fases. En el caso del agua, la curva de coexistencia líquido-vapor termina en el punto crítico, lo que significa que deja de haber dos fases claramente diferenciadas. Podemos visualizar esto pensando que en el punto
para seguir teniendo coexistencia de fases. En el caso del agua, la curva de coexistencia líquido-vapor termina en el punto crítico, lo que significa que deja de haber dos fases claramente diferenciadas. Podemos visualizar esto pensando que en el punto ![]() de la curva de coexistencia que se muestra a continuación, la energía libre de Gibbs posee dos mínimos, tal como habíamos descripto anteriormente. Al acercarnos al punto crítico, como en el punto
de la curva de coexistencia que se muestra a continuación, la energía libre de Gibbs posee dos mínimos, tal como habíamos descripto anteriormente. Al acercarnos al punto crítico, como en el punto ![]() , los mínimos se encuentran muy próximos, o, lo que es equivalente, las propiedades de la fase líquida y la gaseosa se hacen muy similares. Finalmente, en el punto crítico los dos mínimos coinciden, dejando de diferenciarse las propiedades que antes distinguían a las dos fases. La transición deja de ser de primer orden, para convertirse en principio en una de orden superior. Más allá del punto crítico no se aprecian cambios bruscos en las propiedades termodinámicas del sistema.
, los mínimos se encuentran muy próximos, o, lo que es equivalente, las propiedades de la fase líquida y la gaseosa se hacen muy similares. Finalmente, en el punto crítico los dos mínimos coinciden, dejando de diferenciarse las propiedades que antes distinguían a las dos fases. La transición deja de ser de primer orden, para convertirse en principio en una de orden superior. Más allá del punto crítico no se aprecian cambios bruscos en las propiedades termodinámicas del sistema.
|
|
|